Guide pratique : Le syndrome de Turner
Rédigé par Margot COMEL interne en génétique, février 2025
Relecture : Pr GENEVIEVE David, responsable du Centre de référence des anomalies du développement syndromes malformatifs, CHU Montpellier ; Pr Thomas EDOUARD responsable du Centre de référence des maladies endocriniennes rares de la croissance et du développement et le Dr Olivier PATAT, responsable Centre de compétence des anomalies du développement et syndromes malformatifs CHU de Toulouse. Mme MARTIN Danielle déléguée régionale de l’association Association des groupes amitié Turner (AGAT) (ORPHA:881 ; OMIM #313300, #309590)
Qu’est-ce que le syndrome de Turner ?
Le syndrome de Turner a été décrit cliniquement en 1938. Dans la grande majorité des cas, il est dû à l’absence partielle ou totale d’un chromosome X chez une fille. . La formule chromosomique de ces patientes est 45,X. Il est observé chez environ 1 femme sur 2000 à 1 femme sur 2500. Il entraine principalement un retard de croissance et une insuffisance ovarienne précoce.
Caractéristiques cliniques
On retrouve plusieurs formes de syndromes de Turner. En effet, la petite taille et l’insuffisance ovarienne sont assez constants mais les autres symptômes sont variables selon les personnes et le type d’anomalie chromosomique. Les personnes peuvent avoir une forme dite complète et homogène si toutes les cellules de leur corps ont un seul chromosome X. Des formes en mosaïque existent également, c’est-à-dire que certaines cellules de l’individu ont un seul chromosome X et d’autres en ont bien deux. En général, les personnes présentant une monosomie du chromosome X en mosaïque ont une présentation clinique avec moins de symptômes ou une expression des symptômes plus légers que les individus avec une forme complète. Nous détaillerons tout d’abord les signes cliniques habituellement associés aux formes les plus fréquentes (45,X), puis nous reviendrons sur les particularités chromosomiques plus rares et leur impact.
Ce syndrome peut être diagnostiqué pendant la grossesse, en anténatal :
- Sur des signes d’appel échographiques comme l’hyperclarté nucale avec parfois une maladie grave dès la vie fœtale (anasarque) ;
- De manière fortuite comme par exemple si un caryotype est réalisé dans le cadre d’une suspicion d’une autre maladie chromosomique.
Le syndrome de Turner est une maladie qui peut toucher de nombreux organes ou systèmes, cette maladie nécessite donc une prise en charge multidisciplinaire dans des centres spécialisés.
Des malformations cardiaques sont observées chez environ 50% des jeunes filles (les deux plus fréquentes étant la bicuspidie aortique et la coarctation aortique). Des malformations rénales sont observées chez 30 à 70% des nouveau-nées, sans conséquence connues au long terme dans la majorité des cas, mais nécessitant une surveillance si elles sont retrouvées.
A la naissance, les nouveau-nés peuvent avoir un lymphœdème des extrémités et une nuque épaisse, qui disparaissent habituellement durant les premières semaines de vie. Le lymphœdème peut néanmoins réapparaitre plus tard.
Pour les enfants, il existe des courbes de croissance spécifiques. En effet, 95% des filles ayant un syndrome de Turner ont un retard de taille pendant l’enfance et à l’âge adulte (taille adulte moyenne en l’absence de traitement de -3,5 Déviations Standards soit 144 cm).
Dans les formes classiques de syndrome de Turner, il n’y a pas de trouble du développement intellectuel. Cependant, elles peuvent avoir des difficultés dans certains apprentissages (70% des personnes), notamment scolaires (souvent en mathématiques), qui peuvent être prises en charge par des professionnels. On note également un risque plus important d’avoir un Trouble du Spectre de l’Autisme (TSA) chez ces jeunes filles et un risque de Trouble Déficitaire de l’Attention avec ou sans Hyperactivité (TDAH). Des troubles anxieux sont également décrits.
Les otites sont fréquentes chez les petites filles et peuvent entrainer un risque de surdité de transmission. Une surdité neurosensorielle est également fréquente (50% des femmes entre 15 et 35 ans) et s’aggrave avec l’âge.
Les adolescentes ont souvent une insuffisance ovarienne (90%) qui se manifeste par un retard pubertaire (absence de développement des seins après l’âge de 13 ans) et une absence de règles. Un démarrage pubertaire spontané s’observe tout de même chez 30% des jeunes filles (dans les formes incomplètes en mosaïque).
Un surpoids survient souvent à l’adolescence, des mesures diététiques et de l’activité physique sont recommandées de manière préventive.
Une scoliose apparaît chez 10 à 20% des adolescentes.
Les grains de beauté sont souvent nombreux et doivent être surveillés.
Certaines maladies auto-immunes sont plus fréquentes chez les femmes avec un syndrome de Turner comme une dysthyroïdie (hypo ou hyperthyroïdie), une maladie cœliaque ou des maladies inflammatoires du tube digestif. Les femmes atteintes de ce syndrome auraient un risque de 60% au cours de la vie d’avoir une maladie auto-immune.
On note également un risque modérément augmenté d’être diabétique (diabète de type 2) et d’avoir de l’hypertension artérielle (50% des femmes à l’âge adulte). Environ 60% des femmes atteintes ont un taux de cholestérol trop élevé à l’âge adulte, ce qui peut entrainer une stéatose hépatique (35% des personnes avec un syndrome de Turner).
Le risque de dilatation aortique à l’âge adulte implique une surveillance cardiaque régulière à partir de l’adolescence et à l’âge adulte. En effet, la dissection aortique touche 1 à 2% des personnes avec un syndrome de Turner et survient à un âge précoce (en moyenne 30 à 35 ans).
Aspects génétiques
Les cellules humaines comportent 46 chromosomes. On distingue des chromosomes identiques chez les hommes et les femmes, au nombre de 44, classés en 22 paires identiques numérotées de 1 à 22 : ce sont les gonosomes. Ces deux derniers chromosomes distinguent les hommes des femmes : les hommes ont 1 chromosome X et 1 chromosome Y alors que les femmes ont 2 chromosomes X.
Dans la forme la plus fréquente de syndrome de Turner, les femmes ont un seul chromosome X : elles ont donc 45 chromosomes.
La plupart des cas ne sont pas héréditaires, c’est une anomalie chromosomique qui survient de manière aléatoire et accidentelle lors de la formation des cellules reproductrices chez le parent de la personne atteinte.
Cette particularité chromosomique peut être retrouvée dans toutes les cellules du corps : on parle alors de monosomie X (45,X). On peut retrouver un seul chromosome X dans certaines cellules mais pas dans d’autres : on parle alors de composante chromosomique en mosaïque (45,X avec mosaïcisme). Les individus avec un caryotype en mosaïque 45,X/46,XX ont des manifestations cliniques plus légères. On retrouve des malformations cardiaques congénitales du côté gauche. L’obésité et l’hypertension artérielle sont moins fréquentes. L’âge des premières règles est proche de la normale et il y a plus de chance d’obtenir une grossesse spontanée.
Certains types d’anomalies du chromosome X peuvent conduire à un chromosome X non fonctionnel et donc à des symptômes similaires au syndrome de Turner par monosomie X :
- L’isochromosome Xq qui correspond à un chromosome X constitué de 2 copies de son bras long qui sont connectés tête à tête. Les femmes avec un chromosome Xq isocentrique ont une présentation clinique intermédiaire et ont moins de risque d’avoir une dilatation aortique ;
- Le chromosome X en anneau avec une perte des extrémités des bras courts et longs du chromosome. Les personnes avec un chromosome X en anneau ont un risque augmenté de syndrome métabolique. En cas de perte d’une région chromosomique appelée XIST, les troubles cognitifs ont tendance à être plus sévères ;
- Les délétions Xp (du bras court) ou Xq (du bras long) avec une suppression d’une partie du chromosome X.
Dans les formes rares du syndrome de Turner avec une mosaïque 45,X/46,XY, les individus présentent un risque accru de développer des tumeurs gonadiques (notamment des gonadoblastomes), en particulier si les gonades sont situées en position intra-abdominale. Cette spécificité justifie souvent une gonadectomie prophylactique pour prévenir ces tumeurs.
Diagnostic
Le diagnostic est principalement établi lors de la réalisation d’un caryotype et/ou d’une FISH (Hybridation Fluorescente In Situ permettant de détecter la présence ou l’absence des chromosomes X) sur les gonosomes. On retrouve alors un seul chromosome X dans environ 40 à 50% des cas de syndrome de Turner, ou alors une forme en mosaïque (c’est-à-dire que certaines cellules du corps ont un seul chromosome X et que d’autres cellules en ont deux) qui représente environ 50% des cas. Dans 2 à 10% des cas, on retrouve des anomalies sur un des chromosomes X conduisant à un chromosome X non fonctionnel (voire paragraphe ci-dessus).
Le diagnostic peut aussi être réalisé par Analyse Chromosomique sur Puces à ADN (ACPA) ou par séquençage de l’exome (étude du nombre de copie des chromosomes) voire du génome.
En cas de diagnostic anténatal, une confirmation postnatale du caryotype est recommandée.
Figure 1 : Caryotype chez une femme. On visualise tous les chromosomes en double dont 2 chromosomes X. On dit que la formule chromosomique est 46,XX. Image partagée avec courtoisie par le laboratoire de cytogénétique du CHU de Montpellier.
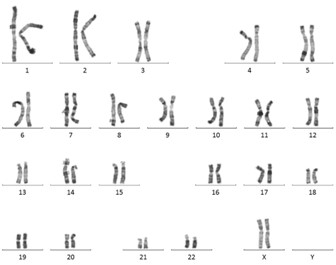
Figure 2 : Caryotype chez une personne avec un syndrome de Turner. On visualise un seul chromosome X : c’est une monosomie X. On dit que la formule chromosomique est 45,X. Image partagée avec courtoisie par le laboratoire de cytogénétique du CHU de Montpellier.
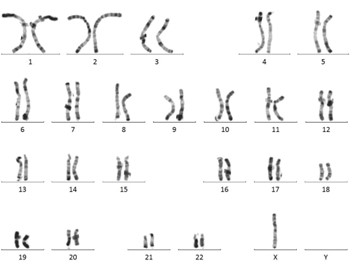
Figure 3 : Caryotype avec un isochromosome Xq (c’est le chromosome X de gauche). Ce chromosome est constitué de 2 bras longs qui sont connectés par leurs têtes. Sur le schéma de droite, les bras bleus représentent les bras longs du chromosome X et les bras oranges sont les bras courts. Image partagée avec courtoisie par le laboratoire de cytogénétique du CHU de Montpellier.
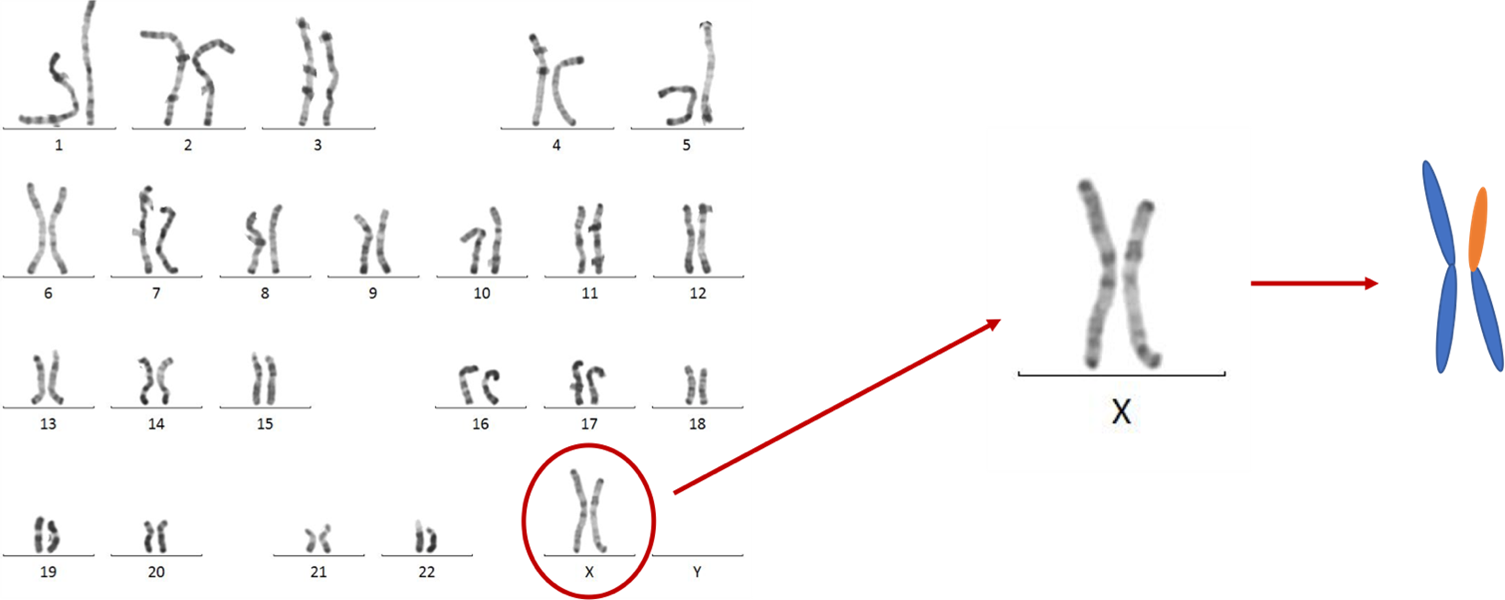
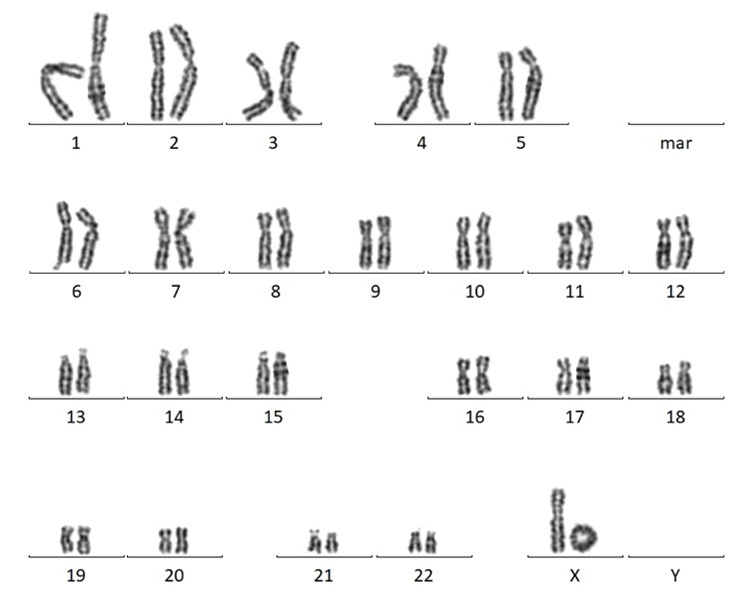
Figure 4 : Caryotype avec un chromosome X en anneau. Ce chromosome a perdu le bout de son bras long et le bout de son bras court, les bras ont ensuite fusionné entre eux pour former un anneau. Image partagée avec courtoisie par le laboratoire de cytogénétique du CHU de Montpellier.
Prise en charge globale, traitement et suivi
Il n’existe pas à ce jour de traitement curatif du syndrome de Turner, la prise en charge est donc symptomatique et préventive. La prise en charge doit se faire dans un Centre Expert Maladies Rares (Centre de Référence Maladies Rares, CRMR, ou Centre de Compétence Maladies Rares, CCMR) du syndrome de Turner qui sont les CRMR/CCMR des anomalies du développement syndromes malformatifs, les CRMR/CCMR des maladies endocriniennes de la croissance et du développement (CRESCENDO) et les CRMR/CCMR des pathologies gynécologiques rares (PGR) au CHU de Montpellier et/ou de Toulouse (coordonnées dans la partie « Professionnels de santé en Occitanie »).
A la naissance et dans la petite enfance, des chirurgies peuvent être nécessaires en fonction de la présence ou non de certaines malformations notamment cardiaques.
L’échographie cardiaque et l’IRM cardiaque sont recommandées au moment du diagnostic pour évaluer la potentielle dilatation de l’aorte, une coarctation aortique ou d’autres malformations cardiaques congénitales. En l’absence de facteurs de risque de dissection aortique, une imagerie de surveillance par échographie ou IRM pour évaluer l’aorte est recommandée tous les 5 ans.
Un traitement par hormone de croissance peut être proposé chez les enfants présentant un ralentissement de la vitesse de croissance et/ou un retard de taille (taille < -2 DS), le plus souvent à partir de l’âge de 3-4 ans quand le diagnostic est fait précocement. Un âge précoce de début de traitement (notamment avant l’âge de la puberté) est associé à une meilleure efficacité. Ce traitement doit être mis en place par un médecin spécialiste (pédiatre endocrinologue) et permet une augmentation moyenne de la taille adulte de +1 à +1,5 DS (5 à 10 cm) avec une variabilité individuelle importante. Des apports optimaux en calcium et en vitamine D doivent être assurés.
A l’adolescence, en cas d’insuffisance ovarienne, un traitement hormonal substitutif par œstrogènes, secondairement associé à des progestatifs, permet le développement pubertaire. Ce traitement est habituellement débuté vers l’âge de 11-12 ans et/ou lorsque l’âge osseux est de 11 ans. Le suivi de ce traitement doit être assuré par un médecin spécialiste (pédiatre endocrinologue). Comme pour toutes les autres jeunes filles, la vaccination contre le papillomavirus est conseillée. En cas de fonction ovarienne résiduelle (forme en mosaïque), une préservation de la fertilité et une contraception doivent être discutées.
Chez l’adulte, le traitement hormonal substitutif oestrogéno-progestatif doit être poursuivi en cas d’insuffisance ovarienne afin de prévenir les risques cardio-vasculaires et osseux. Il a également un impact positif sur les fonctions cognitives et la vie sexuelle.
Un suivi cardiovasculaire est mis en place tout au long de la vie (dans des Centres de Référence ou de Compétence Maladies Rares pour la coordination de la prise en charge si possible). Un traitement antihypertenseur est nécessaire en cas d’hypertension artérielle. En l’absence de facteurs de risque de dissection aortique, une imagerie de surveillance par échographie ou IRM cardiaque est recommandée tous les 10 ans ainsi qu’avant de planifier une grossesse. En cas de dilatation aortique, le suivi est le même que pour toute autre cause de dilatation aortique, soit une imagerie de surveillance de l’aorte au moins une fois par an.
50% des femmes avec un syndrome de Turner ont un surpoids ou une obésité. 25 à 70% de ces femmes auront un diabète de type 2 au cours de leur vie. Une prise en charge nutritionnelle est fortement recommandée. L’activité physique est également recommandée mais sera à adapter en fonction de la présence ou non d’une dilatation de l’aorte. Un traitement par bêtabloquants peut être discuté et prescrit par le cardiologue en fonction du diamètre de la dilatation. Une chirurgie prophylactique de la racine de l’aorte peut être indiquée chez les adolescentes ou les femmes adultes en fonction de l’importance de la dilatation.
L’hypercholestérolémie est fréquente dans le syndrome de Turner et est notamment influencée par l’obésité, le syndrome métabolique et le diabète. Ces facteurs de risque cardiovasculaires augmentent les risques de maladie coronarienne. On retrouve aussi un risque un peu plus élevé de troubles du rythme cardiaque qui justifie un suivi par ECG régulier chez le cardiologue.
Un appareillage auditif est proposé en cas d’hypoacousie.
En cas de réapparition du lymphœdème, l’orientation vers des spécialistes (dans les Centres de Référence ou de Compétence Maladies Rares pour la coordination de la prise en charge) tels que des physiothérapeutes, des ergothérapeutes et des massothérapeutes lymphatiques peut être utile.
Si une préservation de la fertilité a été réalisée (fonction ovarienne résiduelle dans les formes en mosaïque), un parcours AMP peut être réalisée avec les ovocytes conservés. Chez les femmes porteuses d’une anomalie chromosomique en mosaïque, un diagnostic prénatal (DPN) est recommandé en cas de grossesse spontanée.
Si la préservation de la fertilité n’a pas pu être réalisée et en cas de désir de grossesse, les femmes avec un syndrome de Turner peuvent faire appel à un don d’ovocytes auprès d’un centre d’Assistance Médicale à la Procréation (AMP).
Une consultation pré-conceptionnelle est recommandée et une imagerie cardio-aortique est obligatoire dans l’année qui précède la grossesse du fait des dilatations aortiques, de même qu’un bilan hépatique et une glycémie. En effet, les personnes atteintes du syndrome de Turner ont un risque 25 fois plus élevé de faire une dissection de l’aorte durant la grossesse. En cas d’indice de taille aortique > 2,5 cm/m2, une chirurgie prophylactique de l’aorte sera fortement recommandée avant de débuter la grossesse. En cas de grossesse, une surveillance cardio-vasculaire régulière sera mise en place durant toute la grossesse (une échographie cardiaque réalisée toutes les 12 semaines pendant la grossesse en présence d’une dilatation aortique, ou plus fréquemment au cas par cas). En cas d’augmentation rapide du diamètre aortique, une IRM sera demandée. Une évaluation glycémique aura lieu au premier et au deuxième trimestre. Le risque de fausse couche et d’anomalies chromosomiques est augmenté en cas de grossesse spontanée. Pour toute grossesse, les risques de complications cardio-vasculaires, de prématurité et de césarienne sont augmentés.
Une prise en charge psychologique est recommandée. Des comportements d’isolement, d’anxiété et de dépression sont fréquents à l’adolescence et à l’âge adulte. Une évaluation cognitive et neuropsychologique est recommandée dans la prise en charge des personnes avec un syndrome de Turner.
Fiches de suivi
Scolarisation et Insertion Professionnelle
La scolarisation se fait généralement en milieu ordinaire. Une évaluation voire une adaptation scolaire peuvent être nécessaires en cas de troubles des apprentissages. La présence d’un accompagnant des élèves en situation de handicap (AESH) auprès de l’enfant à l’école peut être une aide précieuse en cas de difficultés d’apprentissage et/ou de TDAH.
Si une enfant avec un syndrome de Turner dû à un chromosome X en anneau a de plus grosses difficultés scolaires, une scolarisation en milieu adapté est tout à fait envisageable. À l'âge adulte, il existe un large éventail de situations. Certains ont une bonne insertion sociale, affective et professionnelle. D'autres ont plus de difficultés notamment en ce qui concernent l’insertion professionnelle. Il peut alors être nécessaire de proposer une évaluation neuropsychologique et de la qualité de vie avec des tests spécifiques effectuées par les neuropsychologues, afin de proposer une prise en charge adaptée et des aménagements si nécessaire. Il est possible de proposer l’aide d’un assistant de service social.
Se référer au Guide Emploi et Maladies Rares : https://www.maladies-rares-occitanie.fr/actualites/223-un-nouvel-outil-le-guide-emploi-et-maladie-rare
Suivi médical et paramédical
Le diagnostic et la prise en charge du syndrome de Turner requièrent une approche multidisciplinaire essentielle, impliquant différents professionnels de santé dans des structures spécialisées, comme les Centres de Compétence et les Centres de Référence Maladies Rares.
Dès l’évaluation initiale, la collaboration entre pédiatres, endocrinologues, généticiens, néphrologues, cardiologues et gynécologues est importante pour gérer les potentielles et différentes complications associées (anomalies rénales, cardiaques, hormonales, etc.). À cela s’ajoutent les interventions paramédicales d’un infirmier en éducation thérapeutique, d’un diététicien pour la surveillance nutritionnelle, d’un psychomotricien pour le soutien au développement moteur, d’un orthophoniste pour d’éventuels troubles du langage ainsi que d’un psychologue. Une telle prise en charge globale permet d'optimiser le suivi à long terme et la qualité de vie des personnes.
Accès aux droits
Des aides financières, telle que l’Allocation pour l’Éducation des Enfants en situation de Handicap (AEEH), peuvent également être mises en place pour compenser la prise en charge du handicap. L’AEEH est basée sur les constatations des professionnels médicaux et paramédicaux d’une part, et des aidants principaux d’autre part.
Le médecin référent ou le médecin traitant se charge de remplir la demande de prise en charge à 100% au titre d’une Affection Longue Durée (ALD) hors liste. Il remplit également le certificat médical pour le dossier de la Maison Départementale de l’Autonomie (MDA) (anciennement appelée Maison Départementale des Personnes en situation de Handicap, MDPH).
Enfin, le dispositif régional Maladies Rares Occitanie est également à la disposition des professionnels qui accompagnent les personnes et les familles pour les soutenir dans les différentes démarches sociales : aide à la constitution du dossier pour la Maison Départementale de l’Autonomie (MDA), etc.
Associations de patients
Associations de patientes
- Association des groupes amitié Turner (AGAT) : http://www.agat-turner.org/
- Association Turner et vous : https://www.turneretvous.org/
- Turner Syndrome Foundation (international) : https://turnersyndromefoundation.org/
Références
- Protocole National de Diagnostic et de Soins (PNDS), Haute Autorité de Santé. Syndrome de Turner. https://www.has-sante.fr/jcms/c_632797/fr/syndrome-de-turner
- Shankar Kikkeri N, Nagalli S. Turner Syndrome. 2023 Aug 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan–. PMID: 32119508.
- Gravholt CH, Andersen NH, Christin-Maitre S, Davis SM, Duijnhouwer A, Gawlik A, Maciel-Guerra AT, Gutmark-Little I, Fleischer K, Hong D, Klein KO, Prakash SK, Shankar RK, Sandberg DE, Sas TCJ, Skakkebæk A, Stochholm K, van der Velden JA; International Turner Syndrome Consensus Group; Backeljauw PF. Clinical practice guidelines for the care of girls and women with Turner syndrome. Eur J Endocrinol. 2024 Jun 5;190(6):G53-G151. doi: 10.1093/ejendo/lvae050. PMID: 38748847.
- Yoon SH, Kim GY, Choi GT, Do JT. Organ Abnormalities Caused by Turner Syndrome. Cells. 2023 May 11;12(10):1365. doi: 10.3390/cells12101365. PMID: 37408200; PMCID: PMC10216333.
Professionnels de santé en Occitanie Est
CCMR Centre de Compétences des maladies endocriniennes de la croissance et du développement
Hôpital Arnaud de Villeneuve
Centre de référence des anomalies du développement syndromes malformatifs (pour les formes complexes comme les formes avec anneau de l’X)
Hôpital Arnaud de Villeneuve
34295 Montpellier CEDEX 5
France
Plateforme d’Expertise Maladies rares Montpellier-Nîmes (PEMR)
Hôpital Arnaud de Villeneuve, CHU de Montpellier
34295 Montpellier CEDEX 5
France
Professionnels de santé en Occitanie Ouest
Centre de référence des maladies endocriniennes rares de la croissance et du développement (CRESCENDO)
CHU de Toulouse - Hôpital Purpan
TSA 70034 – 31059 Toulouse Cedex 9
France
Centre de référence des pathologies gynécologiques rares (PGR)
CHU de Toulouse – Hôtel-Dieu Saint-Jacques
TSA 80035 – 31059 Toulouse Cedex 9
France
Centre de compétence des anomalies du développement et syndromes malformatifs
CHU de Toulouse - Hôpital Purpan
TSA 70034 – 31059 Toulouse Cedex 9
France